亲和力(Affinity)
1. Vina Score
1.1. 原理
对配体和受体的假设:
- 不变的
- protonation state and charge distribution of molecules
- rigid, the covalent lengths and angles constant of the receptor
- 可变的
- 配体的部分的共价键可以旋转
- 配体的整体可以旋转和平移
可以拆成两部分,分子间的和(配体)分子内的
全局扰动+局部搜索, 得到若干结合构象,按总能量排序,将总能量最小的构象的配体分子内能量记作 然后计算 Vina 得分
vina 分数取 的最小值。
1.2. 安装
set -e
set -x
rm -rf metrics/vina
# check an mkdir
dir_name="metrics/vina"
if [ ! -d $dir_name ]; then
echo "mkdir $dir_name"
mkdir -p $dir_name
fi
cd $dir_name
# vina
wget https://github.com/ccsb-scripps/AutoDock-Vina/releases/download/v1.2.7/vina_1.2.7_linux_x86_64
chmod 700 vina_1.2.7_linux_x86_64
ln -s $(pwd)/vina_1.2.7_linux_x86_64 vina
pip install vina
# meeko
pip install numpy scipy rdkit
pip install git+https://github.com/zeroDtree/Meeko.git
pip install git+https://github.com/prody/prody
# AutoGrid
git clone https://github.com/ccsb-scripps/AutoGrid
cd AutoGrid
autoreconf -i
mkdir Linux64
cd Linux64
../configure --disable-dependency-tracking
make
cd ../..
# ADFR
wget https://ccsb.scripps.edu/adfr/download/1038/ -O adfr.tar.gz
tar -zxvf adfr.tar.gz
cd ADFRsuite_x86_64Linux_1.0
printf "yes\n" | ./install.sh -d Linux64 -c 0
# molscrub
cd ..
git clone https://github.com/zeroDtree/molscrub.git
pip install -e molscrub
# add_to_path_config $(pwd)/ADFRsuite_x86_64Linux_1.0/Linux64/bin
# add_to_path_config $(pwd)/AutoGrid/Linux64
# add_to_path_config $(pwd)1.3. 使用
https://autodock-vina.readthedocs.io/en/latest/docking_basic.html
1.4. 效果
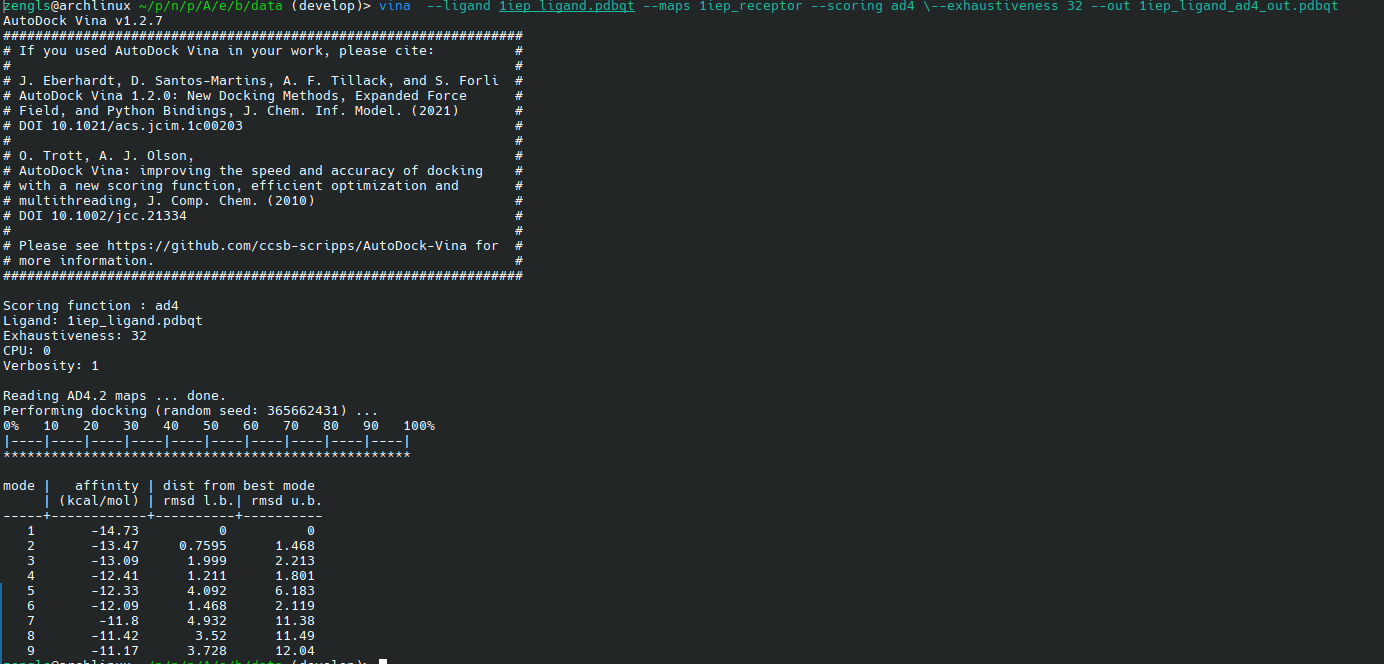
2. UniGSSA
2.1. 原理
计算配体受体互作前后体系的自由能变化。
2.2. 安装流程
依赖:gmxMMPBSA
wget https://valdes-tresanco-ms.github.io/gmx_MMPBSA/dev/env.yml
conda env create --file env.yml
conda activate gmxMMPBSAunigbsa
pip install unigbsa lickit2.3. 使用
unigbsa-pipeline -i example/1ceb/1ceb_protein.pdb -l example/1ceb/1ceb_ligand.sdf -o BindingEnergy.csv2.4. 效果
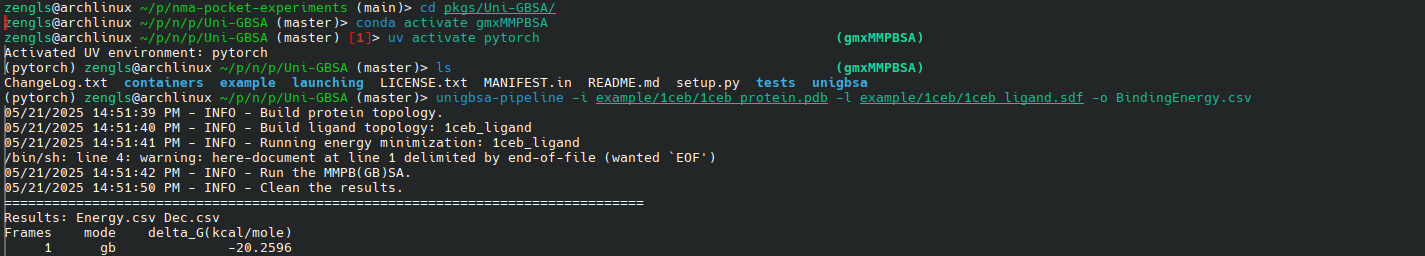
结构的有效性(自一致性)
3. pLDDT
AlphaFold 或 ESMFold 的一个输出头输出的分数
The pLDDT score reflects the confidence in structural predictions on a scale from 0 to 100, with higher scores indicating greater confidence
from transformers import AutoTokenizer, EsmForProteinFolding
model = EsmForProteinFolding.from_pretrained("facebook/esmfold_v1")
tokenizer = AutoTokenizer.from_pretrained("facebook/esmfold_v1")
inputs = tokenizer(["MLKNVQVQLV"], return_tensors="pt", add_special_tokens=False) # A tiny random peptide
outputs = model(**inputs)
plddt = outputs.plddt4. scRMSD
self-consistency root mean square deviation
检查设计出的结构能否自洽地被当前的结构预测工具所再现。
- 使用模型生成一个蛋白质结构
- 然后用 ProteinMPNN 设计出氨基酸序列
- 再用结构预测工具(如 AlphaFold2 或 ESMFold)从这个序列反推出结构,
- 最后比对原始生成结构与预测结构的一致性。
import torch
from torch import Tensor
def rmsd(predicted: Tensor, ground_truth: Tensor, mask: Tensor, eps: float = 1e-10)->Tensor:
"""Calculate RMSD between two protein structures.
Args:
predicted (``Tensor`` shape of ``(..., n_res, 3)``): CA coordinates of predicted protein structure
ground_truth (``Tensor`` shape of ``(..., n_res, 3)``): CA coordinates of ground truth protein structure
mask (``Tensor`` shape of ``(..., n_res)``): mask of valid CA atoms
eps (``float``, *optional*): small value to avoid division by zero. Defaults to 1e-10.
Returns:
``Tensor`` shape of ``(..., )``: RMSD
"""
assert predicted.shape == ground_truth.shape # (bs, n, 3)
assert predicted.shape[-1] == 3
assert mask.shape == predicted.shape[:-1] # (bs, n)
sq_errors = (predicted - ground_truth) ** 2 * mask.unsqueeze(-1)
mse = torch.sum(sq_errors) / (torch.sum(mask) * 3)
rmsd = torch.sqrt(mse.clamp_min(eps))
return rmsd
if __name__ == "__main__":
predicted = torch.tensor([[1.3, 7.2, 1.5], [4.0, 2.9, -1.7], [1.2, 4.2, 4.3]])
ground_truth = torch.tensor([[2.3, 7.4, 1.5], [4.0, 2.7, -1.7], [1.2, 4.2, 4.3]])
mask = torch.tensor([1, 1, 1])
print(rmsd(predicted, ground_truth, mask))5. scTM
self-consistency Template Modeling score
可使用tmtools计算TM-score
pip install tmtoolsimport numpy as np
from torch import Tensor
from tmtools import tm_align
import torch
def get_tm_score(coords1: Tensor, coords2: Tensor) -> Tensor:
"""Calculate TM-score between two protein structures.
Args:
coords1 (``Tensor`` shape of ``(n_res, 3)``): CA coordinates of protein 1
coords2 (``Tensor`` shape of ``(n_res, 3)``): CA coordinates of protein 2
Returns:
``Tensor`` shape of ``(, )``: TM-score
"""
assert coords1.ndim == 2 and coords2.ndim == 2 and coords1.shape == coords2.shape
assert coords1.shape[1] == 3 and coords2.shape[1] == 3
seq_len = coords1.shape[0]
coords1 = coords1.cpu().numpy()
coords2 = coords2.cpu().numpy()
seq = "A" * seq_len
res = tm_align(coords1, coords2, seq, seq)
return torch.tensor(res.tm_norm_chain1)
if __name__ == "__main__":
coords1 = torch.tensor([[1.3, 7.2, 1.5], [4.0, 2.9, -1.7], [1.2, 4.2, 4.3]])
coords2 = torch.tensor([[2.3, 7.4, 1.5], [4.0, 2.7, -1.7], [1.2, 4.2, 4.3]])
print(get_tm_score(coords1, coords2))